Protein
iPhoPred
:Predicting the Phosphorylation Sites in Human Protein
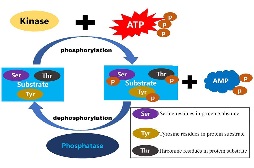 |
iPhoPred is a server for the prediction of phosphorylation sites based on machine learning method. In the process of training model, position score function and the correlation information of physicochemcial properties between two residues were considered to build feature sets, and then analysis of variance (ANOVA) was proposed to exclude noise and redundant information. The incremental feature selection (IFS) was used to determine the optimal number of feature which could produce the maximum auROC. Jackknife test results show that the proposed method can discriminate phosphothreonine (T) with auROC of 99.0%, classify phosphotyrosine (Y) with auROC of 99.2% and predict phosphorylated serine (S) with auROC of 90.4%, respectively. We hope the webserver could provide convenience for wet-experimental scholars. |
HBPred2.0
:Identification of hormone-binding protein
 |
HBPred 2.0 is a webserver for the identification of hormone-binding protein (HBP). In the predictor, protein sequences were coded by tripeptide composition method and binomial distribution method. An overall accuracy of 97.15% was obtained based on SVM in the 5-fold cross-validation test. Besides, area under the ROC curve was 0.994, which indicated that the proposed model is very powerful and deserves our trust. We hope the webserver could provide convenience for Biochemistry scholars. |
ApoliPred
:Identification of apolipoproteins
 |
The web-server ApoliPred was developed to identify the apolipoproteins based on the sequence information. The analysis of variance was used to seek optimized dipeptide composition. The anticipated overall success rates are 98.4% by using five-fold cross-validation. 96.2% apolipoproteins and 99.3% non-immunoglobulins can be correctly identified. |
C2Pred
:Prediction of cell-penetrating peptides
 |
The web-server C2Pred was developed to identify the cell-penetrating peptides (CPPs) based on the sequence information. The analysis of variance was used to seek optimized dipeptide composition. The anticipated overall success rates are 83.6% by using 5-fold cross-validation. 81.5% CPPs and 85.6% non-CPPs can be correctly identified. |
IGPred
:Identification of immunoglobulins
 |
The web-server IGPred was developed to identify the immunoglobulins (antibodies) based on the sequence information. The analysis of variance was used to seek optimized dipeptide composition. The anticipated overall success rates are 96.9% by using jackknife cross-validation. 96.3% immunoglobulins and 97.5% non-immunoglobulins can be correctly identified. |
iACP
: Predicting anticancer peptide
 |
The web-server iACP was developed to identify the anticancer peptides based on the sequence information. The wrapper-type feature selection technique was used to seek optimized g-gap dipeptide. The overall accuracy of 95.06% was obtained in the jackknife cross-validation. |
AodPred
: Predicting the antioxidant proteins
 |
The tool AodPred was developed to identify antioxidant proteins based on the sequence information. The feature selection technique was used to seek optimized g-gap dipeptide. AodPred achived an accuracy of 74.79% for antioxidant predictions in the jackknife cross-validation test. 75.09% antioxidant proteins and 74.48% non-antioxidant proteins were correctly identified. |
CaLecPred
: Predicting the cancerlectins
 |
The web-server CalecPred was developed to identify cancerlectin based on the sequence information. The analysis of variance was used to seek optimal g-gap dipeptide. The anticipated overall success rates are 75.19% by using jackknife cross-validation. |
JPred
: Predicting the types of J-proteins
 |
The web-server was developed to identify the types of J-proteins by using reduced amino acid alphabet. The overall accuracy was over 94% by using jackknife cross-validation. |
 |
Heat shock proteins (HSPs) are a type of functionally related proteins present in all living organisms, both prokaryotes and eukaryotes. They play essential roles in protein–protein interactions such as folding and assisting in the establishment of proper protein conformation and prevention of unwanted protein aggregation. Their dysfunction may cause various life-threatening disorders, such as Parkinson’s, Alzheimer’s, and cardiovascular diseases. Based on their functions, HSPs are usually classified into six families: (i) HSP20 or sHSP, (ii) HSP40 or J-class proteins, (iii) HSP60 or GroEL/ES, (iv) HSP70, (v) HSP90, and (vi) HSP100. Although considerable progress has been achieved in discriminating HSPs from other proteins, it is still a big challenge to identify HSPs among their six different functional types according to their sequence information alone. With the avalanche of protein sequences generated in the post-genomic age, it is highly desirable to develop a high-throughput computational tool in this regard. To take up such a challenge, a predictor called iHSP-PseRAAAC has been developed by incorporating the reduced amino acid alphabet information into the general form of pseudo amino acid composition. One of the remarkable advantages of introducing the reduced amino acid alphabet is being able to avoid the notorious dimension disaster or overfitting problem in statistical prediction. It was observed that the overall success rate achieved by iHSP-PseRAAAC in identifying the functional types of HSPs among the aforementioned six types was more than 87%, which was derived by the jackknife test on a stringent benchmark dataset in which none of HSPs included has P40% pairwise sequence identity to any other in the same subset. It has not escaped our notice that the reduced amino acid alphabet approach can also be used to investigate other protein classification problems. |
subGolgi
: Prediction of subGolgi locations of proteins
 |
The web-server subGolgi was developed to identify Golgi-resident proteins and their types based on the sequence information. The ANOVA was used to find out optimized dipeptide. The anticipated overall success rates are 85.4% in discriminating between cis-Golgi proteins and trans-Golgi proteins and 79.9% in the prediction of Golgi-resident proteins by using jackknife cross-validation. |
TetraMito
: Prediction of submitochondria locations of proteins
 |
The web-server TetraMito was developed to identify submitochondria location of proteins based on the sequence information. The binomial distribution was used to find out optimized tetrapeptide. The anticipated overall success rates are 91.1% in predicting inner membrane, matrix and outer membrane proteins by using jackknife cross-validation. |
ChloPred
: Prediction of subchloroplast locations of proteins
 |
The web-server ChloPred was developed to identify subchloroplast location of proteins based on the sequence information. The binomial distribution was used to find out optimized tripeptide. The anticipated overall success rates are 88.03% by using jackknife cross-validation. 92.21% envelope proteins, 93.20% thylakoid membrane, 52.63% thylakoid lumen and 85.00% stroma can be correctly identified. |
AcalPred
: Discriminating acidic enzymes from alkaline enzymes
 |
The web-server AcalPred was developed to discriminate between acidic and alkaline enzymes based on the sequence information. The analysis of variance was used to seek optimized g-gap dipeptide. The anticipated overall success rates are 96.7% by using jackknife cross-validation. 96.3% acidic enzymes and 97.1% alkaline enzymes can be correctly identified. |
ThermoPred
: Identification of thermophilic proteins
 |
The web-server ThermoPred was developed to identify thermophilic proteins based on the sequence information. The 93.8% thermophilic proteins and 92.7% non-thermophilic proteins can be correctly predicted by use of jackknife cross-validation. |
iVKC-OTC
: Identifying the subfamilies of voltage-gated potassium channe
 |
The web-server iVKC-OTC was developed to predict the subfamilies of votaged-gated potassium channel using tripeptide compositions. The binomial distribution was proposed to optimize tripeptides for eliminating the redundant features and improving the efficiency and performance of prediction. In the jackknife cross-validation, the proposed method achieved an overall accuracy of 96.77% with 93.92% average sensitivity, which are superior to that of other three state-of-the-art classifiers. |
iCTX-Type
: Predicting the types of conotoxins in targeting ion channels
 |
The web-server iCTX-Type was developed to predict the types of conotoxins in targeting ion channels. The overall accuracy of 91.07% was achieved by using jackknife cross-validation. |
IonchanPred 2.0
: Identification of ion channels and their types
 |
IonchanPred 2.0 is a server for the prediction of ion channel and their types based on machine learning method. In the process of training model, dipeptide frequency and the correlation information of physicochemcial properties between two residues were considered to build feature sets, and then analysis of variance (ANOVA) was proposed to exclude noise and redundant information. The incremental feature selection (IFS) was used to determine the optimal number of feature which could produce the maximum accuracy. Jackknife cross-validated results show that the proposed method can discriminate ion channels from non-ion channels with an overall accuracy of 87.8%, classify voltagegated ion channels and ligand-gated ion channels with an overall accuracy of 94.0% and predict four types (potassium, sodium, calcium and anion) of voltage-gated ion channels with an overall accuracy of 92.6%, respectively. We hope the webserver could provide convenience for wet-experimental scholars. |
Lypred
: Prediction of bacterial cell wall lyase
 |
The web-server Lypred was developed to predict Bacterial Cell Wall Lyase via pseudo amino acid composition. The average accuracy of 84.82% with the auROC of 0.926 was achieved in jackknife cross-validation. |
PHYPred
: Prediction of bacteriophage enzymes and hydrolases
 |
The web-server PHYPred was developed to identify the phage enzymes and hydrolases based on the sequence information. The analysis of variance was used to seek optimized g-gap dipeptide. In jackknife cross-validation, our method can discriminate the bacteriophage enzymes from non-enzymes with maximum overall accuracy of 84.3% and can further discriminate phage hydrolase from non-hydrolases with maximum overall accuracy of 93.5%. |
PHPred (version1.0)
and
PHPred (version2.0)
: Prediction of bacteriophage proteins located in host cell
 |
PHPred 2.0 is an online server for the prediction of phage proteins located in the host cell (PH proteins) and their subcellular localizations based on machine learning method. In the process of training model, separated dipeptide compositions and the descriptor CTD of physicochemical properties were considered to build feature sets, and then analysis of variance (ANOVA) was proposed to exclude noise and redundant information. The incremental feature selection (IFS) was used to determine the optimal number of feature which could produce the maximum accuracy. Results of jack-knife test by SVM show that the proposed method can discriminate PH proteins from non-PH proteins with an overall accuracy of 86.7%, classify phage proteins located in the host cell membrane (PHM proteins) and phage proteins located in the host cell cytoplasm (PHC proteins) with an overall accuracy of 97.9%, respectively. We hope the web server could provide convenience for wet-experimental scholars. |
PVPred
: Identification of phage virion proteins
 |
The bacteriophage virion proteins play extremely important roles in the fate of host bacterial cells. Accurate identification of bacteriophage virion proteins is very important for understanding their functions and clarifying the lysis mechanism of bacterial cells. In this study, a new sequence-based method was developed to identify phage virion proteins. In the new method, the protein sequences were initially formulated by the g-gap dipeptide compositions. Subsequently, the analysis of variance (ANOVA) with incremental feature selection (IFS) was used to search for the optimal feature set. It was observed that, in jackknife cross-validation, the optimal feature set including 160 optimized features can produce the maximum accuracy of 85.02%. By performing feature analysis, we found that the correlation between two amino acids with one gap was more important than other correlations for phage virion protein prediction and that some of the 1-gap dipeptides were important and mainly contributed to the virion protein prediction. This analysis will provide novel insights into the function of phage virion proteins. |
Mycosub: Predicting subcellular localization of mycobacterial proteins
 |
The web-server MycoSub was used to predict the subcellular localizations of mycobacterial proteins based on optimal tripeptide compositions. We achieved overall accuracy of 89.71% with average accuracy of 81.12% in jackknife cross-validation. |
MycoSec: Identification of mycobacterial secretory proteins
 |
The web-server MycoSec was developed to predict secretory proteins in mycobacterium tuberculosis with pseudo amino acid composition. The averaged accuracy of 87.18% with the AUC of 0.93 was achieved in jackknife cross-validation. |
MycoMemSVM: Identification of mycobacterial membrane proteins and their types
 |
The web-server MycoMemSVM was developed to identify mycobacterial membrane proteins and their types based on the over represented tripeptide compositions. The binomial distribution was used to find out over represented tripeptide. The anticipated overall success rates are 93.0% in discriminating between mycobacterial membrane proteins and mycobcaterial non-membrane proteins and 93.1% in the prediction of mycobacterial membrane protein types by using jackknife cross-validation. |