RNA
XG-PseU:an eXtreme Gradient Boosting based method for identifying pseudouridine sites
 |
As one of the most popular post-transcriptional modifications, pseudouridine (Ψ) participates in a series of biological processes. Therefore, the efficient detection of pseudouridine sites is very important in revealing its functions in biological processes. Although experimental techniques have been proposed for identifying Ψ sites at single-base resolution, they are still labor intensive and expensive. Recently, to fill the experimental method's gap, computational methods have been proposed for identifying Ψ sites. However, their performances are still unsatisfactory. In this paper, we proposed an eXtreme Gradient Boosting (xgboost)-based method, called XG-PseU, to identify Ψ sites based on the optimal features obtained using the forward feature selection together with increment feature selection method. Our results demonstrated that XG-PseU is superior or at least complementary to existing methods for identifying pseudouridine sites. |
iMRM:is a predictor for identifying post-transcriptional modification sites
 |
RNA modifications play critical roles in a series of cellular and developmental processes, such as RNA localization and degradation, dynamic changes in RNA structure , RNA localization and degradation , RNA splicing , circadian rhythm , etc. Accordingly, it is very useful to accurately identify the location information of RNA modification sites. In this article we developed a new predictor called iMRM, which is able to simultaneously identify m6A, m5C, m1A, ψ and A-to-I modifications in Homo sapiens, Mus musculus and Saccharomyces cerevisiae. In iMRM, the feature selection technique was used to pick out the optimal features. The results from both 10-fold cross validation and jackknife test demonstrated that the performance of iMRM is superior to existing methods for identifying RNA modifications. |
 |
N6-methyladenosine (m6A) is the most abundant post-transcriptional modification and involves a series of important biological processes. Therefore, accurate detection of the m6A site is very important for revealing its biological functions and impacts on diseases. Although both experimental and computational methods have been proposed for identifying m6A sites, few of them are able to detect m6A sites in different tissues. With the consideration of the spatial specificity of m6A modification, it is necessary to develop methods able to detect the m6A site in different tissues. In this work, by using the convolutional neural network (CNN), we proposed a new method, called im6A-TS-CNN, that can identify m6A sites in brain, liver, kidney, heart and testis of Homo sapiens, Mus musculus and Rattus norvegicus. In im6A-TS-CNN, the samples were encoded by using the one-hot encoding scheme. The results from both a 5-fold cross-validation test and independent dataset test demonstrate that im6A-TS-CNN is better than the existing method for the same purpose. |
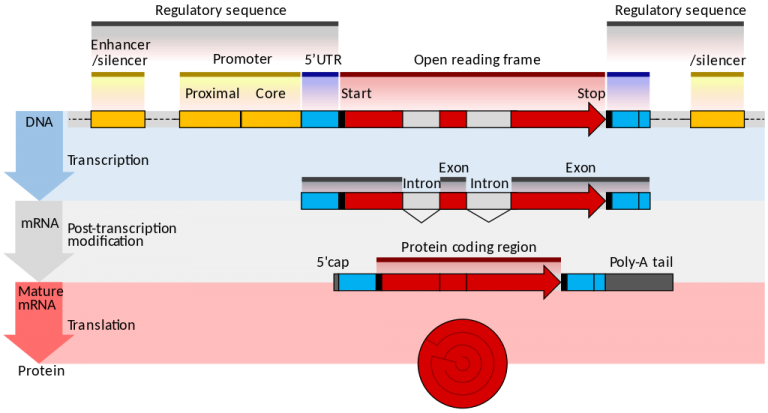 |
One of the first steps for functional genomic annotation is promoter identification. The promoter region is located near the transcription start sites and regulates transcription initiation of the gene. Promoter of protein-coding genes are gradually being well understood, yet no machine learning based method exist for the promoter of non-coding RNA (ncRNA) genes of human and mouse. Since experimental methods are cost and ineffective, developing efficient and accurate computational tools are necessary.
|
iRNA-Methyl
,
m6Apred
,
MethyRNA
,
M6ATH
and
iRNA(m6A)-PseDNC
Identification of M6A sites in RNAs
 |
Occurring at adenine (A) with the consensus motif GAC, N6-methyladenosine (m6A) is one of the most abundant modifications in RNA, which plays very important roles in many biological processes. The nonuniform distribution of m6A sites across the genome implies that, for better understanding the regulatory mechanism of m6A, it is indispensable to characterize its sites in a genome-wide scope. Although a series of experimental technologies have been developed in this regard, they are both timeconsuming and expensive. With the avalanche of RNA sequences generated in the postgenomic age, it is highly desired to develop computational methods to timely identify their m6A sites. In view of this, a predictor called “iRNA-Methyl” is proposed by formulating RNA sequences with the “pseudo dinucleotide composition” into which three RNA physiochemical properties were incorporated. Rigorous crossvalidation tests have indicated that iRNA-Methyl holds very high potential to become a useful tool for genome analysis. |
iRNA-PseU: Identifying RNA pseudouridine sites
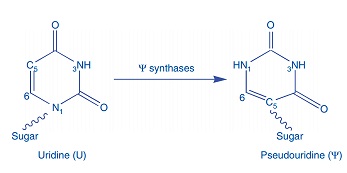 |
As the most abundant RNA modification, pseudouridine plays important roles in many biological processes. Occurring at the uridine site and catalyzed by pseudouridine synthase, the modification has been observed in nearly all kinds of RNA, including transfer RNA, messenger RNA, small nuclear or nucleolar RNA, and ribosomal RNA. Accordingly, its importance to basic research and drug development is self-evident. Despite some experimental technologies have been developed to detect the pseudouridine sites, they are both time-consuming and expensive. Facing the explosive growth of RNA sequences in the postgenomic age, we are challenged to address the problem by computational approaches: For an uncharacterized RNA sequence, can we predict which of its uridine sites can be modified as pseudouridine and which ones cannot? Here a predictor called “iRNA-PseU” was proposed by incorporating the chemical properties of nucleotides and their occurrence frequency density distributions into the general form of pseudo nucleotide composition (PseKNC). It has been demonstrated via the rigorous jackknife test, independent dataset test, and practical genome-wide analysis that the proposed predictor remarkably outperforms its counterpart. |
PAI
and
iRNA-AI
: Predicting adenosine to inosine editing sites
 |
The web-server was developed to identify the Adenosine to Inosine editing sites in the D. melanogaster transcriptome. |
iRNA-PseColl
: Identifying m1A, m6A, and m5C sites in human
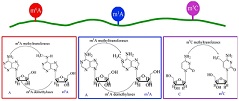 |
The web-server was developed to identify RNA modifications in H. sapiens transcriptome. At present, the N1-methyladenosine (m1A), N6-methyladenosine(m6A) and 5-methylcytosine modifications (m5C) can be identified by using the current platform. |
iRNA-3typeA
: Identifying m1A, m6A, and A-to-I sites in human and mouse
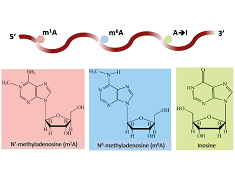 |
The web-server allows for simultaneously identifying the most frequently observed N1-methyladenosine (m1A), N6-methyladenosine (m6A) and adenosine to inosine (A-to-I) modifications in both H. sapiens and M. musculus transcriptomes. |
iRNA-2OM
: Identifying 2'-O-Methylation Sites in human
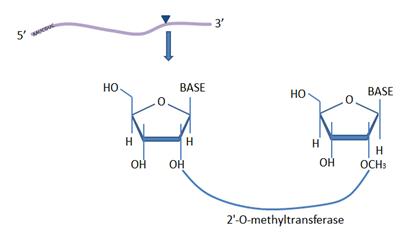 |
2'-O-methylation is catalyzed by the 2'-O-methylationtransferase, in which a methylation group is added to the 2' hydroxyl group of the ribose moiety of a nucleotide. iRNA-2OM is a system for identifying whether a RNA sequence contains the 2'-O-methylation site based on machine learning method. In the process of training model, nucleotide chemical properties were considered to build feature sets. The mRMR was used to determine the optimal number of feature, thus to achieve the optimal training model. 5-fold show that the proposed method can identifying the 2'-O-methylation site with an overall accuracy of 97.95%, We hope the webserver could provide convenience for wet-experimental scholars. |
iRNAD
: identifying D modification sites
 |
Dihydrouridine commonly called 8U or D for short is one of the single most common form of RNA post-transcriptional modifications. D modification was formed by adding two hydrogen atoms to a uridine (U) base of which the carbon-carbon double bond at positions 5 and 6 are reduced. The modification can promote the conformational flexibility of individual nucleotide bases. And its levels are increased in cancerous tissues. Therefore, detecting D modification in RNA will contribute not only to molecular biology but also to medical science. iRNAD is a system for identifying whether a RNA sequence contains D modification sites based on machine learning method. In the process of training model, the RNA samples derived from five species were encoded by nucleotide chemical property and nucleotide density. Support vector machine was utilized to perform classification. The final model could produce the overall accuracy of 96.18% with the area under the receiver operating characteristic curve of 0.9839 in jackknife cross-validation test. We hope the webserver could provide convenience for wet-experimental scholars. |
iRNA-m5C
: identifying m5C modification sites
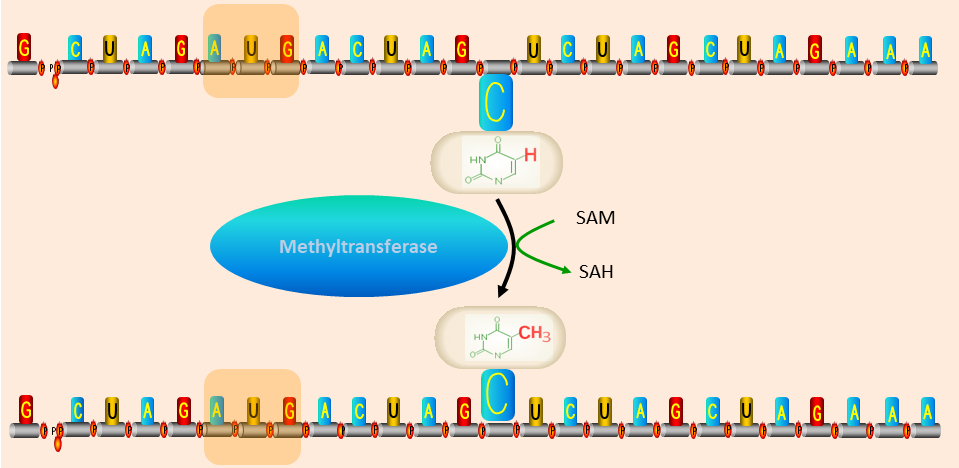 |
5-methylcytosine (m5C) is formed by transferring methyl groups to the 5-th position of the cytosine ring catalyzed by RNA methyltransferases. Accurate identification of m5C site is a key step in understanding its biological functions. In this work, we constructed a more powerful and reliable model for identifying m5C sites. To train the model, we collected experimentally confirmed m5C data from Homo Sapiens (H. Sapiens), Mus musculus (M. musculus), Saccharomyces cerevisiae (S. cerevisiae) and Arabidopsis thaliana (A. thaliana), and compared the performances of different feature extraction methods and classification algorithms for optimizing prediction model. Based on the optimal model, a novel predictor called iRNA-m5C was developed for the recognition of m5C sites. Finally, we critically evaluated the performance of iRNA-m5C and compared it with existing methods. The result showed that iRNA-m5C could produce the best prediction performance. We anticipate that the iRNA-m5C will become a powerful tool for large scale identification of m5C sites. |
iRNA-m7G
: identifying m7G modification sites
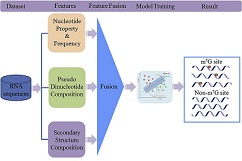 |
N7-methylguanosine (m7G) is an essential RNA modification and participates in a series of biological processes. However, our knowledge about its biological functions is very limited. In order to reveal its new functions, it is necessary to develop various methods to identify m7G sites. Since experimental methods to detect m7G sites are still expensive, we proposed the first computational method, called iRNA-m7G, to detect m7G sites in Homo Sapiens. iRNA-m7G is developed by fusing multiple features, i.e., nucleotide property & frequency, pseudo dinucleotide composition, and secondary structure composition. The framework of iRNA-m7G is shown as following. |
iRNA-m2G
: identifying m2G modification sites
 |
At present N2-methylguanosine (m2G) was only found in tRNA, the knowledge about its biological functions is very limited. In order to reveal its new functions and to detect whether m2G exists in other types of RNA, it is necessary to develop various methods to identify m2G sites. Considering the fact that there is no high throughout experimental methods to detect m2G sites, we developed iRNAm2G, the first computational method, to identify m2G sites. |
iRNA-m6A
: identifying m6A modification sites in multiple tissues of mammals
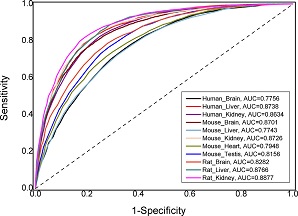 |
N6-methyladenosine (m6A) is the methylation of the adenosine at the nitrogen-6 position, which is the most abundant RNA methylation modification and involves a series of important biological processes. Accurate identification of m6A sites in genome-wide is invaluable for better understanding their biological functions. In this work, an ensemble predictor named iRNA-m6A was established to identify m6A sites in multiple tissues of human, mouse and rat based on the data from high-throughput sequencing techniques. In the proposed predictor, RNA sequences were encoded by physical-chemical property matrix, mono-nucleotide binary encoding and nucleotide chemical property. Subsequently, these features were optimized by using minimum Redundancy Maximum Relevance (mRMR) feature selection method. Based on the optimal feature subset, the best m6A classification models were trained by Support Vector Machine (SVM) with 5-fold cross-validation test. Prediction results on independent dataset showed that our proposed method could produce the excellent generalization ability. |
 |
Long non-coding RNAs (lncRNAs) are a class of RNA molecules with more than 200 bases. They have important functions in cell development and metabolism, including genetic markers, genome rearrangements, chromatin modifications, cell cycle regulation, transcription and Translation, etc. Generally, their functions are also closely related to their location in a cell. The aberrant expression of lncRNAs is associated with several types of cancer, Alzheimer's disease, etc. The study on their localization could provide preliminary insight into their cellular functions. In this work, we designed a sequence-based predictor called “iLoc-LncRNA” to predict the subcellular locations of LncRNAs. In the predictor, a high-quality benchmark dataset including four locations was constructed based on the RNALocate database. The key octonucleotide (8-tuple) features were incorporated into the general PseKNC (Pseudo K-tuple Nucleotide Composition) via the binomial distribution approach to formulate lncRNA samples. The support vector machine (SVM) was adopted to perform discrimination. In rigorous 5-fold cross-validations, we achieved the maximum overall accuracy of 86.72%, suggesting that the proposed predictor is promising and will provide important guidance in this area. |
iLoc-mRNA : Predicting the subcellular location of mRNAs in human
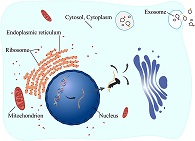 |
The locating process of mRNA might provide spatial and temporal regulation of mRNA and protein functions. The iLoc-mRNA is an excellent predictor which could forecast the subcellular location of the mRNA to four main location: cytosol/cytoplasm, ribosome, endoplasmic reticulum and nucle-us/exosome/dendrite/mitochondrion. iLoc-mRNA was built based on Support Vector Meachine and achieved the overall accuracy of 90.12%. |